眼优角蚱两性成体转录组差异分析
覃英灿,容万韬,李月妹,陆桂红,李晓东
(河池学院化学与生物工程学院,广西宜州 546300)
蚱类昆虫在分类上属于直翅目Orthoptera蝗亚目Caelifera蚱总科Tetrigoidea,由于其背板呈菱形,旧称菱蝗。蚱类昆虫为不完全变态昆虫,在整个昆虫纲中属于较为原始的类群,约在2.2亿年前,由一个共同祖先进化而来(Songetal.,2015)。大多数种类分布于热带和亚热带,是生物多样性和动物区系的重要组成部分,世界已知 9科 270属1 880多种,我国已知7科57属788种,占世界三分之一多(邓维安,2016)。由于蚱类昆虫既不是益虫也不是害虫,所以国内外对蚱类昆虫的研究不多,主要集中在分类学方面,其它方面较少。但近年来发现,由于蚱类昆虫多以苔藓、地衣或腐殖质为食,属地栖性昆虫,运动能力不强,与环境特征联系紧密,可以作为重要的生态指示物种(邓维安等,2007;Tsuruietal.,2010;Wennerstenetal.,2012)。因此有必要采用新的研究技术,对这一古老的类群进行多方面的研究,以挖掘该类群的科学和应用方面的研究价值。
眼优角蚱Eucriotettixoculatus属于刺翼蚱科Scelimenidae优角蚱属Eucriotettix,广泛分布于中国南部各省及南亚地区,属于蚱类中的广布和常见种类(郑哲民,2005),具有代表性。本研究基于转录组高通量测序技术,对眼优角蚱E.oculatus雌雄两性虫体之间的转录差异进行研究,以从转录组层面了解蚱总科昆虫雌雄分化的分子基础,为昆虫分子生物学、发育生物学和进化生物学提供一定的基础资料。
1.1 材料准备
为了尽可能避免不同环境因素和发育时期引起的雌、雄虫之间转录组的差异,本研究所用的眼优角蚱是2019年6-7月分别在广西河池市宜州区和南丹县不同地点采集的雌性和雄性成虫带回实验室交配产卵后,取其卵块在昆虫饲养室中孵化并在稳定的环境条件下从1龄饲养到成虫后收集的进入成虫期时间相近的雌、雄成虫样品。雌虫样品组(编号AF)由9个生物学重复样品(编号为AF1-AF9)组成,且每个生物学重复样品均由3头雌虫组成。雄虫样品组(编号AM)也是由9个生物学重复样品(编号为AM1-AM9)组成,每个样品也由3头雄虫组成。所有样品均为活虫经48 h饥饿后用液氮冻存。
1.2 转录组测序
使用QIAGEN动物组织RNA提取试剂盒提取各样品总RNA并构建cDNA文库,然后基于Illumina技术测序平台,采用双末端测序(Pair-End)法,完成18个样品二代转录组测序分析。
1.3 基因差异表达分析
本研究测定的所有二代转录组数据与本实验室前期已测定的眼优角蚱三代转录组数据(由卵和各龄期雌、雄虫所组成的混合样品经Pacbio Sequel II技术测序平台利用单分子实时测序法获得的三代数据,编号为EO)联合分析。首先使用二代转录组数据对三代转录组数据进行转录本的校正,然后利用CD-HIT软件(Fuetal.,2012)对校正后的consensus序列进行去冗余,接着对去冗余的序列基于Nr(Lietal.,2002),Nt,Pfam(Finnetal.,2008),KOG/COG(Tatusovetal.,2003),Swiss-prot(Amosetal.,2000),KEGG(Kanehisaetal.,2004),GO(Ashburneretal.,2000)共7种数据库进行基因功能注释。同时以去冗余后得到的转录本作为基因的参考序列(ref),将Illumina测序得到的二代转录组测序数据中每个样品的clean reads比对到ref上。利用RSEM软件(Lietal.,2011)对比对结果进行统计,进一步得到每个样品比对到每个基因上的readcount值,并对其进行FPKM转换。基于基因表达的RPKM值,使用DEseq差异表达基因鉴定程序计算AF和AM样品组之间表达量不同的基因。差异基因筛选标准为|log2 (FoldChange)|>0和qvalue<0.05。
1.4 差异基因表达的GO和KEGG分析
采用GOseq(Youngetal.,2010)和KOBAS软件(Chenetal.,2011)对差异基因集进行GO功能富集分析,KEGG通路富集分析。富集分析基于超几何分布原理,其中差异基因集为差异显著分析所得差异基因并注释到GO或KEGG数据库的基因集,背景基因集为所有进行差异显著分析的基因并注释到GO或KEGG数据库的基因集。富集分析结果是对差异比较组合的所有差异基因集、上调基因集、下调基因集进行富集。
1.5 实时荧光定量PCR验证
从眼优角蚱雌性和雄性转录组中筛选出的差异表达基因中随机挑取9个,以β-actin作为内参基因,采用实时荧光定量PCR (qRT-PCR)的方法对转录组的准确性进行验证(验证样品与转录组测序样品一致),验证基因和对应的引物序列见表1。使用反转录试剂盒将总RNA反转录合成cDNA,反转录体系为40 μL(冰上配制):5×PrimeScriptRTMaster Mix (Perfect Real-time) 8 μL和RNA模板2 000 ng,最后加入RNase-free dH2O补全至40 μL。反应条件为:37°C 15 min,85°C 5 s,置于4℃冰箱保存。采用ChamQTM SYBR®qPCR Master Mix染料试剂盒进行实时荧光定量PCR扩增,PCR反应体系20 μL:SYBR Premix Ex Taq (2×) 10 μL,10 μmol/L上、下游引物各1 μL,cDNA模板2 μL,ddH2O补全至20 μL。PCR反应程序为:95℃预变性30 s;
95℃变性5 s,60℃退火34 s,40个循环。

表1 实时荧光定量引物Table 1 Primer sequences for Real-time quantitative PCR
2.1 转录本校正和去冗余
通过本研究测定的二代转录组数据对三代转录组数据校正后,三代consensus的碱基数目有所提升,其它指标基本无变化(表2),说明眼优角蚱转录组二代和三代数据整体质量较高。进一步按照序列之间95%的相似性对校正后的转录本序列进行聚类去冗余,最终得到13 432条transcripts,总长度26 977 000 bp,平均长度2 009 bp,最大长度8 297 bp,N50长度远大于1 000 bp(表3),说明测序效果较好,可以进行后续分析。

表2 转录本校正前后长度分布统计表Table 2 Statistical table of length distribution of transcripts before and after correction

表3 去冗余后序列长度分布统计表Table 3 Statistical table of sequence length distribution after de-redundancy
2.2 基因功能注释
对去冗余之后的序列进行基因功能注释,通过七大数据库共有12 074条transcripts被注释,占整个transcripts的90%,未成功注释的transcripts仅占比10%,说明注释成功率比较高(图1)。对Nr数据库所获得的基因注释与已公布的基因组信息的物种进行匹配,其中被匹配最多的物种有20个(图2),分别是内华达古白蚁Zootermopsisnevadensis、亚利桑那沫蝉Clastopteraarizonana、飞蝗Locustamigratoria、麦茎蜂Cephuscinctus、赤拟谷盗Triboliumcastaneum等皆为昆虫纲物种,因此可证明本研究转录组注释结果的可靠性。

图1 七大数据库注释结果统计图Fig.1 Statistical chart of annotation results of seven databases
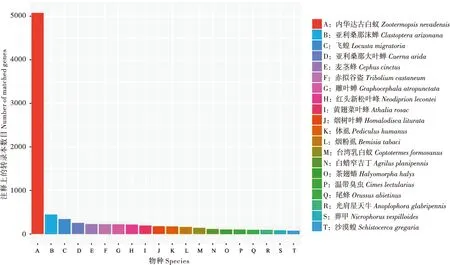
图2 Nr数据库物种注释统计图Fig.2 Statistical chart of species annotation in NR database
2.3 差异表达基因筛选
为了获得雌性和雄性眼优角蚱的表达量不同的基因,运用FPKM法对雌性组(AF)和雄性组(AM)的transcripts进行分析,共获得两者之间的差异表达基因3 597个,其中雌性组(AF)相对于雄性组(AM),共获得1 295个显著上调基因,获得2 302个显著下调基因(图3)。

图3 眼优角蚱雌性与雄性间差异表达基因的比较Fig.3 Comparison of differential expressed genes between male and female Eucriotettix oculatus
2.4 差异表达基因GO富集结果
对差异表达基因进行GO功能分析可以了解差异表达基因的生物学意义。差异表达基因GO功能分类显示,共有2 344条transcripts被GO注释,并被显著富集到生物学过程(Biological process)、细胞组分(Cellular component)和分子功能(Molecular function)三大类的主要35个亚类中。其中细胞成分组织或合成(Cellular component organization or biogenesis)、细胞成分组织(Cellular component organization)、细胞器组织(Organelle organization)、细胞(Cell)、细胞成分(Cell part)、细胞内(Intracellular)、细胞内成分(Intracellular part)、有机环状化合物结合(Organic cyclic compound binding)、杂环化合物结合(Heterocyclic compound binding)、核酸结合(Nucleic acid binding)分别是三大类中显著富集差异基因数量最多的条目(图4)。
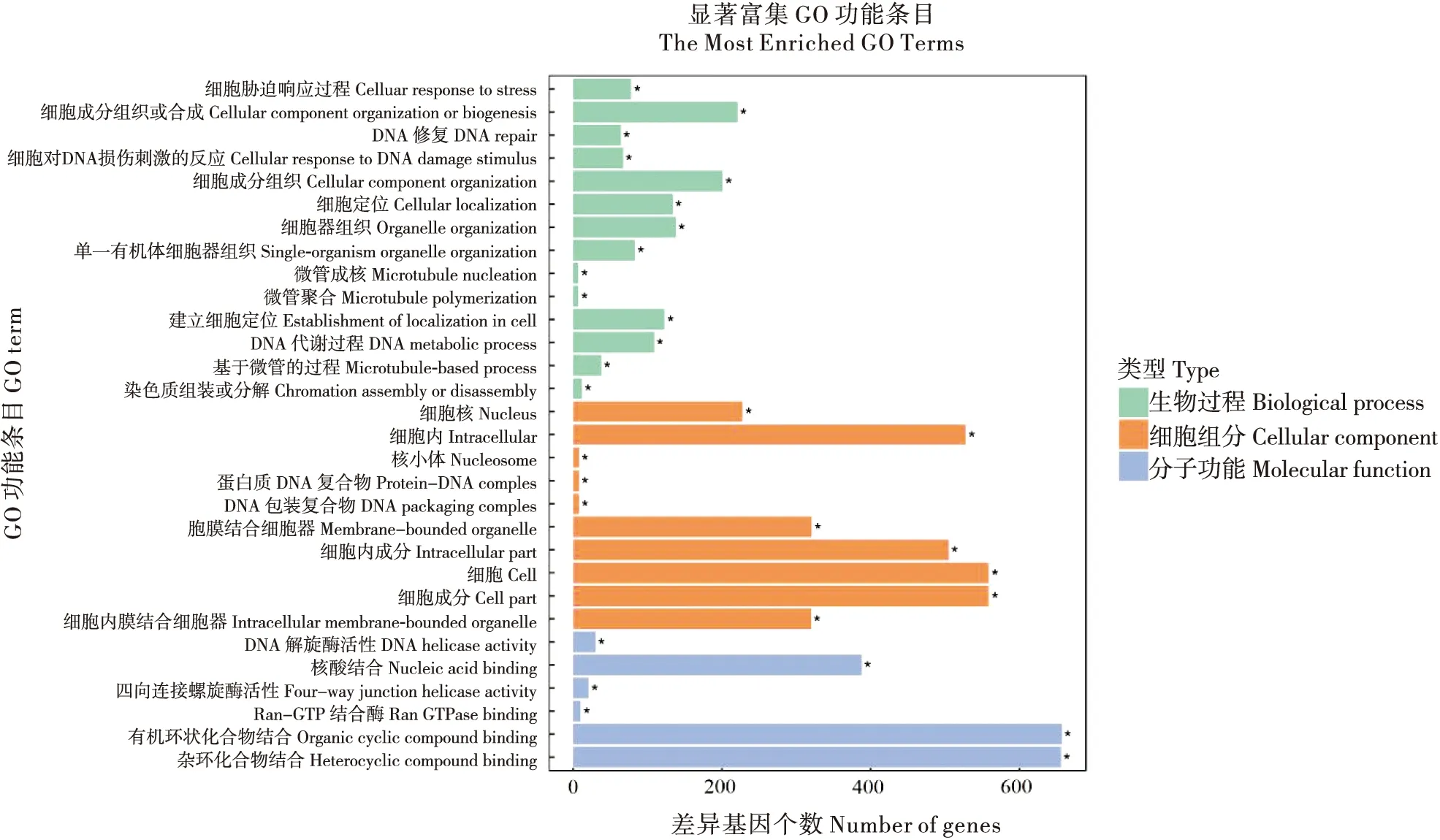
图4 差异表达基因GO功能分类Fig.4 Go functional classification of differentially expressed genes
雌性相对于雄性,表达上调的1 295个基因中,被GO注释的有922个,但没有显著富集的亚类。表达下调的2 302个基因中,被GO注释的有1 422个,显著富集到三大类的47个亚类。其中富集差异基因数目最多的是分子功能(Molecular function)大类中的蛋白质结合(Protein binding)亚类的468个。其次是细胞组分(Cellular component)大类中的细胞器组分(Organelle part)和细胞内细胞器组成(Intracellular organelle part),分别是157和155个。再次是生物过程(Biological process)中的细胞成分组织或生物发生(Cellular component organization or biogenesis)和细胞成分组织(Cellular component organization),分别是140个和133个(图5)。其余亚类富集到的基因数据均在100以下。

图5 下调表达基因GO功能分类Fig.5 Go functional classification of down regulated expressed genes
2.5 差异表达基因KEGG富集结果
雌性相对于雄性,表达上调的1 295个基因经过KEGG注释,共有1 188个基因被注释到248条通路,富集显著的前20条通路(图6),其中神经活性配体受体相互作用(Neuroactive ligand-receptor interaction)、DNA复制(DNA replication)、蛋白质消化吸收(Protein digestion and absorption)、错配修复(Mismatch repair)、嘧啶代谢(Pyrimidine metabolism)和胰腺分泌(Pancreatic secretion)通路是富集最显著的6条通路(qvalue<0.05)。表达下调的2 303个基因中共有1 886个基因被注释到264条通路,富集显著的前20条通路(图7),其中蛋白酶体(Proteasome)、氮代谢(Nitrogen metabolism)、泛素介导的蛋白水解(Ubiquitin mediated proteolysis)、糖酵解/糖异生(Glycolysis/Gluconeogenesis)、丙酮酸代谢(Pyruvate metabolism)和RNA转运(RNA transport)通路是富集最显著的6条通路(qvalue<0.05)。

图6 雌性相对于雄性上调基因KEGG富集散点图Fig.6 Scatter plot of KEGG enrichment in females relative to males of up-regulated gene
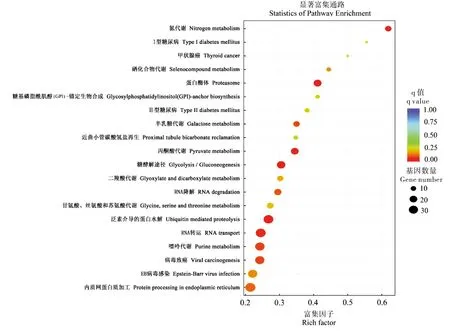
图7 雌性相对于雄性下调基因KEGG富集散点图Fig.7 Scatter plot of KEGG enrichment in females relative to males of down-regulated gene
2.6 实时荧光定量PCR验证结果
qRT-PCR验证结果显示,上调表达的5个基因(VgR、VG2、PRSS1_2_3、TUBA、PLIN2)在转录组测序中的表达均是呈上调趋势,下调表达的4个基因(gdhA、PKLR、UBE2O、CA)在转录组测序中的表达均是呈下调趋势,其中gdhA和PKLR在qRT-PCR验证中均是下调1倍(图8)。
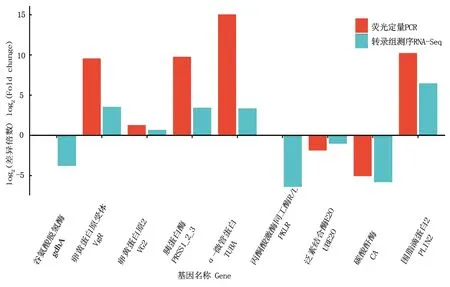
图8 眼优角蚱雌性与雄性差异表达基因qRT-PCR与RNA-Seq分析结果Fig.8 Results of qRT-PCR and RNA-Seq analysis of differential expressed genes between male and female Eucriotettix oculatus
动物的雌雄分化是进化的产物。昆虫作为生物多样性最丰富的动物类群,大多数种类在形态上都表现出了性别二态性,即同种昆虫的雌、雄个体除生殖器官的结构差异外,在大小、颜色、结构等方面也常有明显差异(王孟卿等,2005)。但是,目前对昆虫雌雄性别在转录组层面的差异了解还不充分。
眼优角蚱雌、雄成虫在外部形态上也具有明显的差异,雌虫腹部末端具有细长的产卵瓣,体型较雄虫粗大,前胸背板背面较雄性粗糙,其它构造与体色与雄虫相似(邓维安等,2007)。作为昆虫中较古老的物种,对眼优角蚱雌性和雄性成虫的转录差异开展研究,可为揭示昆虫雌雄差异的分子机制提供一定的基础数据。本研究测定了2组18个样品的二代转录组,并与本实验室已测定的三代转录本进行校正后,最终得到13 432条转录本,平均长度2 009 bp,最大长度8 297 bp,N50长度远大于1 000 bp,说明本研究所得到的转录本质量较较高。本研究仅有10%的转录本没有注释成功,主要是因为数据库中有关直翅目昆虫的基因信息匮乏。
本研究对眼优角蚱雌性和雄性的转录组进行了基因差异表达分析,共获得1 295个显著上调和2 302个显著下调基因。使用qRT-PCR方法验证了9个基因的表达情况,虽然qRT-PCR结果与mRNA测序结果在倍数之间有稍许差异,但是每个基因有同样的上下调趋势,可以说明转录组数据分析的可靠性。在上调的基因中有与雌性发育有关的卵黄蛋白原受体VgR、卵黄蛋白原2VG2、胰蛋白酶PRSS1_2_3、围脂滴蛋白2PLIN2等基因,在下调的基因中有与雄性发育有关的碳酸酐酶CA等基因,说明了差异表达基因与性别紧密联系。为了解差异基因的生物学意义,通过差异基因的GO富集可以发现,差异基因显著富集的GO条目大多是与细胞的形成有关,特别是雄性,其相对于雌性来说,结合(Binding)、细胞组分(Cellular component)和细胞过程(Cellular processes)等条目相关基因显著高表达,说明雌、雄个体在成虫后都有大量的细胞增殖活动,雄性较雌性更加剧烈,这应该是与雄性进入成虫期后要形成更多的精子为交配繁殖做好准备有关(刘杰等,2020)。同时,基于整个基因组背景,应用超几何检验对差异基因进行了的KEGG Pathway富集分析,发现在雌性成虫体内高表达的基因显著富集在主要的6条通路中。其中神经活性配体受体相互作用(Neuroactive ligand-receptor interaction)通路属于生殖相关通路,该通路的基因能影响促性腺素合成和释放,在性腺发育过程中主要是分别作用于脑和卵巢(储明星等,2009;Xuetal.,2015;Zhangetal.,2019)。蛋白质消化吸收(Protein digestion and absorption)和胰腺分泌(Pancreatic secretion)通路属于消化系统,说明雌性眼优角蚱相对雄性消化能力更强,可能是由于雌性个体较大,需要消化吸收更多的营养物质,或者是雌性要储备更多营养物质以备后期生殖需要。而DNA复制(DNA replication)、错配修复(Mismatch repair)和嘧啶代谢(Pyrimidine metabolism)通路均与遗传物质的复制和修复有关。雄性体内高表达的基因也是显著富集在主要的6条通路。其中蛋白酶体(Proteasome)通路可能与雄性眼优角蚱的生精过程有关,因为已有研究发现敲除蛋白酶体REGγ基因可导致精原干细胞数目减少,使雄性小鼠生殖能力降低(Alietal.,2013;李宁等,2016)。氮源是昆虫生长发育和繁殖所必需的营养物质之一。由于食物中氮含量与蛋白质含量直接相关,食物中的氮能直接影响昆虫的交配、取食速率和繁殖等(张碧尧,2019),研究发现与氮代谢有关的精氨酸对精子发生发挥关键作用(Moralesetal.,2003;O’Flahertyetal.,2004;徐学玉等,2017),因此雄性眼优角蚱氮代谢通路被富集。泛素介导的蛋白水解(Ubiquitin mediated proteolysis)属于信号通路,参与卵发生、精子发生和性腺成熟等一系列生殖过程的调控活动(Mengetal.,2015;Pengetal.,2015;Yangetal.,2016;董忠典等,2021)。与能量代谢相关的糖酵解(Glycolysis / Gluconeogenesis)、丙酮酸代谢(Pyruvate metabolism)通路在雄性中被显著富集,可能是雄性眼优角蚱在交配前较雌性活动多,需要消耗更多的能量(肖舒晴等,2019)。RNA转运(RNA transport)通路显著富集,可能是由于蛋白质合成与RNA转运密切相关,雄性需要大量合成与精子形成有关的蛋白,以完成精子储备,这与雄性按蚊转录表达相似(刘杰等,2020)。综上所述,眼优角蚱雌性和雄性成虫在转录水平的差异主要还是集中在与生殖细胞形成和生殖活动有关的基因表达方面。本研究对进一步深入解析昆虫雌雄分化的分子调控差异提供了一定的理论基础。
猜你喜欢 差异基因雌性雄性 为什么萤火虫会发光小学阅读指南·低年级版(2020年9期)2020-10-12基于RNA 测序研究人参二醇对大鼠心血管内皮细胞基因表达的影响 (正文见第26 页)心电与循环(2020年1期)2020-02-27海马是由爸爸的肚里出世阅读与作文(小学低年级版)(2019年11期)2019-12-26被做了手脚的“结婚礼物”文萃报·周五版(2019年18期)2019-09-10五月的“雪”儿童故事画报·自然探秘(2017年7期)2018-03-14紫檀芪处理对酿酒酵母基因组表达变化的影响江苏农业科学(2017年5期)2017-04-15萌物飞碟探索(2016年5期)2016-05-10大豆异黄酮和DEHP对雌性大鼠体内雌激素水平的影响癌变·畸变·突变(2015年3期)2015-02-27SSH技术在丝状真菌功能基因筛选中的应用湖北农业科学(2014年3期)2014-07-21肾阳虚证骨关节炎温针疗效的差异基因表达谱研究云南中医学院学报(2011年4期)2011-07-31- 范文大全
- 说说大全
- 学习资料
- 语录
- 生肖
- 解梦
- 十二星座
-
主题党日活动交流发言8篇
主题党日活动交流发言8篇主题党日活动交流发言篇13月13日,东城区党史学习教育动员大会召开。市委
【活动总结】 日期:2022-12-23
-
2022年4月主题党日活动记录范文15篇
2022年4月主题党日活动记录范文15篇2022年4月主题党日活动记录范文篇1一个崇尚阅读的民族,必然精神饱满、意气风发、活力四射。习近平总书记强调:“学习
【活动总结】 日期:2022-08-01
-
家乡赋|最美的家乡赋
家乡赋 孙传志 今安康市,白河双丰镇,吾之家乡也。三环沃土,山水环抱。其北依山,山系五岭,山
【调研报告】 日期:2020-04-01
-
【人教版1-6年级数学上册知识点精编】1-6年级数学人教版教材
人教版二年级数学上册知识点汇总第一单元长度单位一、米和厘米1、测量物体的长度时,要用统一的标准去测量
【调研报告】 日期:2020-11-08
-
党支部1-12月全年主题党日活动计划表
2022年党支部主题党日活动计划表序号活动时间活动方式活动内容12022年1月专题学习研讨集中观看2022年新年贺词,积极开展学习研讨交流。组织生活会组织党员认真对照党章...
【活动总结】 日期:2022-10-14
-
2022年2月份主题党日活动记录5篇
2022年2月份主题党日活动记录5篇2022年2月份主题党日活动记录篇1尊敬的党组织:在今年的开学初,本人积极参加教研室组织的教研活动,在学校教研员的指
【活动总结】 日期:2022-08-12
-
2023年平安校园建设方案13篇
平安校园建设方案“平安校园”创建工作,我们幼儿园全体教职员工一直把它当作头等大事来抓。领导高度重视,以“平安校园”创建活动为抓手,建立和规范校园安全工作机制
【规章制度】 日期:2023-11-02
-
医院最佳主题党日活动11篇
医院最佳主题党日活动11篇医院最佳主题党日活动篇1 医院最佳主题党日活动篇2为隆重纪念中国共产党成立100周年,进一步巩固党的群众路线教育实践活动成果,切实
【活动总结】 日期:2022-10-29
-
主题党日活动记录202210篇
主题党日活动记录202210篇主题党日活动记录2022篇12021年是中国共产党成立100周年,为广泛开展爱国主义宣传教育,铭记党的历史,讴歌党的光辉历程,
【活动总结】 日期:2022-08-02
-
南京大屠杀国家公祭日悼念文案句子11篇
南京大屠杀国家公祭日悼念文案精选句子1、惟有民魂是值得宝贵的,惟有他发扬起来,中国才有真进步。——鲁迅2、我爱我的祖国,爱我的人民,离开了它,离开了他们,我
【企划文案】 日期:2023-10-20
-
正式的晚宴邀请函 公司晚宴邀请函
尊敬的先生 女士: 我公司谨定于xxxx年xx月xx日xx:xx在xxxx店隆重举行xx市xx届xxxx晚宴(宴会地址:xx区xx路xxxx) 敬请届时光临!xxxxxx集团股份有限公司xxxx有限公司敬邀xxxx年xx月xx日
【简历资料】 日期:2019-08-03
-
《国行公祭,为佑世界和平》课文原文阅读_国行公祭为佑世界和平每段段意
国行公祭,为佑世界和平钟声“国行公祭,法立典章。铸兹宝鼎,祀我国殇。”侵华日军南京大屠杀遇难同胞纪念
【简历资料】 日期:2020-11-28
-
一年级新学期目标简短_一年级学生新学期打算
新学期到了,我是一年级下册的小学生了。 上课的时候,我要认真学习,不做小动作,认真听讲。我要认真学习,天天向上,努力学习,耳朵要听老师讲课,眼睛要瞪得大大的看老...
【简历资料】 日期:2019-10-26
-
[信访复查复核制度作用探讨]信访复查复核有用吗
作为我国特有的一项制度,信访制度的出现并长期存在不是偶然的,虽然一些法学专家认为信访制度具有“人治”
【职场指南】 日期:2020-02-16
-
[党员干部2019年主题教育个人问题检视清单及整改措施2篇] 党员干部
2019年主题教育问题检视清单及整改措施根据主题教育领导小组办公室《关于认真做好主题教育检视问题整改
【求职简历】 日期:2019-11-08
-
网络维护工作内容_(精华)国家开放大学电大专科《网络系统管理与维护》形考任务1答案
国家开放大学电大专科《网络系统管理与维护》形考任务1答案形考任务1理解上网行为管理软件的功能【实训目
【职场指南】 日期:2020-07-17
-
党委会与局长办公会的区别_局长办公会制度
为进一步加强xxx局工作的规范化、制度化建设,提高行政效能,规范议事程序,特制定本制度。一、会议形式1、局长办公会议由局长、副局长参加。由局长召集和主持。根据工作需要...
【求职简历】 日期:2019-07-30
-
《铁拳砸碎“黑警伞”》警示教育片观后感
影片深刻剖析了广西北海市公安局海西派出所原所长张枭杰蜕变堕落的轨迹。观看警示教育片后,做为一名党员教
【简历资料】 日期:2020-08-17
-
如何凝心聚力谋发展【坚定信心谋发展凝心聚力促跨越】
当前,清河正处于在苏北实现赶超跨越基础上全面腾飞的战略机遇期,处于在全市率先实现全面小康基础上率先实
【简历资料】 日期:2020-03-17
-
学生会组织部部长竞选稿5篇
学生会组织部部长竞选稿以“三制”为统领推进农村党的建设中共**市委组织部近年来,**市认真落实中央、省和徐州市委的部署,积极适应发展要求,从加强领导体制、运
【求职简历】 日期:2023-11-06
-
毛概社会实践关于新农村_暑期新农村建设社会实践个人总结
这些文字,无法全部描述这段时间我的所见,也无法完全准确表达我真正想传达的,但它记下了这段时间的点点滴滴,一些记忆犹新的过去。眼中兰田兰田村的天然和淳朴是显而易见的...
【其他范文】 日期:2019-07-10
-
语文评课发言稿2篇_语文评课发言稿
各位领导、各位老师:刚才我们仔细聆听了三位老师按照新课程标准执教的公开课。我个人的感觉是形式多样,风格各异,各具特色,精彩粉呈。大致说来,三位老师执教的观摩课有如...
【汇报体会】 日期:2019-08-02
-
基于合金注入与大视域图像技术的致密储层孔隙与喉道表征
唐相路,萧汉敏,姜振学,刘学伟,杨再权,刘格,张帆1 中国石油大学(北京)油气资源与探测国家重点实验
【其他范文】 日期:2023-06-30
-
【审计局综治情况汇报】
审计局综治情况汇报一年来,我局社会治安综合治理工作坚持以理论和“三个代表”重要思想为指导,在县委、县
【礼仪】 日期:2021-11-12
-
XX镇废旧农膜回收利用工作实施方案7篇
XX镇废旧农膜回收利用工作实施方案7篇XX镇废旧农膜回收利用工作实施方案篇1不埋怨谁,不嘲笑谁,也不羡慕谁,阳光下灿烂,风雨中奔跑,做自己的梦,走自己的路。
【其他范文】 日期:2022-08-18
-
全区居委会换届工作情况报告
下面是小编为大家整理的全区居委会换届工作情况报告文章
【其他范文】 日期:2022-07-31
-
《揭秘科学》读后感
当前位置:>>>2021-10-03现在科技发达,出产书的种类、玩的花样、不同品牌的电脑、手机等等都很多。最近我看了一本书叫《揭秘科学》,里面讲述了天体:太阳是一个不断爆发的巨...
【其他范文】 日期:2022-09-27
-
大学生返家乡社会实践活动方案4篇
大学生返家乡社会实践活动方案4篇大学生返家乡社会实践活动方案篇1时光如白驹过隙,转瞬即逝。为期一个月的“返家乡”大学生社会实践也将迎来了结束。在这短短的一个
【其他范文】 日期:2022-11-02
-
红松森林浴
◎张国中在平原上住得久了,就有了一个奇怪的梦想:如果拥有一片大森林多好,让我远离尘嚣,在大森林里过一
【其他范文】 日期:2023-02-23
-
开发区年轻干部选拔培育工作汇报
近年来,**经开区坚持以习近平新时代中国特色社会主义思想为指导,聚焦新时代干部队伍建设总要求,把年轻干部选拔培育作为一项重要工作,积极健全年轻干部工作常态化机制,不...
【其他范文】 日期:2023-09-23
-
军转座谈会交流发言4篇
军转座谈会交流发言4篇军转座谈会交流发言篇1大家好,我叫贺丽,2015届选调生,来自康定市委组织部,现在省委编办跟班学习。今天,非常荣幸向大家汇报我的学习收
【发言稿】 日期:2022-10-27
-
12岁生日小寿星发言4篇
12岁生日小寿星发言4篇12岁生日小寿星发言篇1各位来宾、各位朋友:大家好!今天,我们欢聚在这里,共同庆祝**十二周岁生日。首先,我代表**的父母以
【发言稿】 日期:2022-07-31
-
党内警告处分表态发言14篇
党内警告处分表态发言14篇党内警告处分表态发言篇1尊敬的各位领导、同事们:大家上午好!刚才会上宣布了党委关于我任职的决定,我首先衷心感谢党委的信任和
【发言稿】 日期:2022-09-13
-
党内警告处分党员讨论发言3篇
党内警告处分党员讨论发言3篇党内警告处分党员讨论发言篇1大家好!作为新时期的一名大学生,认真学习、深刻领会、全面贯彻省党代会精神,是当前和今后一个时期重
【发言稿】 日期:2022-08-07
-
廉政大会总结发言稿7篇
廉政大会总结发言稿7篇廉政大会总结发言稿篇1各位领导,同志们:根据会议安排,我就党风廉政建设工作做表态发言,不妥之处,请批评指正。一、提高认识,切实
【发言稿】 日期:2022-10-30
-
被约谈的表态发言8篇
被约谈的表态发言8篇被约谈的表态发言篇1各位领导、各位党员大家好:这天我能站在鲜红的党旗下,
【发言稿】 日期:2022-12-24
-
破冰提能大讨论个人发言4篇
破冰提能大讨论个人发言4篇破冰提能大讨论个人发言篇1党史学习教育开展以来,我坚持读原著、学原文、悟原理。今天,根据会议安排,现在我就“学史明理”主题谈几点个
【发言稿】 日期:2022-10-09
-
巡察整改专题民主生活会总结发言8篇
巡察整改专题民主生活会总结发言8篇巡察整改专题民主生活会总结发言篇1按照区委统一部署和纪监委、巡察办关于召开党史学习教育专题组织生活会的工作安排,近期我紧贴
【发言稿】 日期:2022-10-12
-
我最敬佩的人开头_我敬佩的一个人作文20篇2020年
我敬佩的一个人作文20篇 我敬佩的一个人作文一): 我身边有很多值得我们敬佩的人,但我最敬佩的一
【发言稿】 日期:2020-11-10
-
纪委书记工作表态发言4篇
纪委书记工作表态发言4篇纪委书记工作表态发言篇1在镇党委政府正确领导下,在全村干部和群众的共同努力下,紧紧围绕建设社会主义新农村工作为重点,尽职尽责,与时俱
【发言稿】 日期:2022-09-30
-
学习周永开先进事迹心得体会3篇
学习周永开先进事迹心得体会【一】通过学习周永开老先生先进事迹后,结合自己工作思考,感慨万千。同样作为
【格言】 日期:2021-04-10
-
最满意的三项工作200字【最新党办公务员副主任提拔考察个人三年思想工作总结报告】
党办公务员个人三年工作总结近三年来,本人在组织、领导的关心指导和同事们的团结协作下,尽快完成主角的转
【格言】 日期:2021-02-26
-
XX老干局推进党建与业务深度融合发展工作情况调研报告:党建调研报告
XX老干局推进党建与业务深度融合 发展工作情况的调研报告 党建工作与业务工作融合发展始终是一个充满生
【成语大全】 日期:2020-08-28
-
中国共产党第三代中央领导集体的卓越贡献
中国共产党第三代中央领导集体的卓越贡献 --------------继往开来铸就辉煌 【摘要】改
【成语大全】 日期:2020-03-20
-
信息技术2.0能力点 [全国中小学教师信息技术应用能力提升工程试题题库及参考答案「精编」]
全国中小学教师信息技术应用能力提升工程试题题库及答案(复习资料)一、判断题题库(A为正确,B为错误)
【格言】 日期:2020-11-17
-
党建工作运行机制内容有哪些_构建基层党建工作运行机制探讨
党的基层组织是党在社会基层组织中的战斗堡垒,是党的全部工作和战斗力的基础。加强和改进县级以下各类党的
【经典阅读】 日期:2020-01-22
-
电大现代教育原理_最新国家开放大学电大《现代教育原理》形考任务2试题及答案
最新国家开放大学电大《现代教育原理》形考任务2试题及答案形考任务二一、多项选择题(共17道试题,共3
【成语大全】 日期:2020-07-20
-
集合推理_七,推理与集合
七推理与集合1 期中考试数学成绩出来了,三个好朋友分别考了88分,92分,95分。他们分别考了多少分
【名人名言】 日期:2020-12-18
-
基层党务工作基本内容_党建基本工作有哪些
党建基本工作有哪些(一) 基层党建工作包括哪些内容 选择了大学生村官这条路,你就与农村基层党
【名人名言】 日期:2020-08-06
-
2023年中国行政区划调整方案(设想优秀3篇
中国行政区划调整方案(设想优秀民政部第二次行政区划研讨会会议内容一、缩省的意义与原则1.意义1)利于减少中间层次中国行政区划层级之多为世界之最,既使管理成本
【周公解梦】 日期:2024-02-20
-
关于三农工作重要论述心得体会3篇
关于三农工作重要论述心得体会3篇关于三农工作重要论述心得体会篇1习近平总书记指出:“建设现代化国家离不开农业农村现代化,要继续巩固脱贫攻坚成果,扎实推进乡村
【学习心得体会】 日期:2022-10-29
-
【福生庄隧道坍塌处理方案】 福生庄隧道在哪里
(呼和浩特铁路局大包电气化改造工程指挥部,内蒙古呼和浩特010050)摘要:文章介绍了福生庄隧道
【学习心得体会】 日期:2020-03-05
-
五个一百工程阅读心得体会13篇
五个一百工程阅读心得体会13篇五个一百工程阅读心得体会篇1凡益之道,与时偕行。在全国网络安全和信
【学习心得体会】 日期:2022-12-07
-
城管系统警示教育心得体会9篇
城管系统警示教育心得体会9篇城管系统警示教育心得体会篇1各党支部要召开多种形式的庆七一座谈会,组织广大党员进行座谈,回顾党的光辉历程,畅谈党的丰功伟绩,
【学习心得体会】 日期:2022-10-09
-
发展对象培训主要内容10篇
发展对象培训主要内容10篇发展对象培训主要内容篇1怀着无比激动的心情,我有幸参加了__新区区委党校20__年第四期(区级机关)党员发展对象培训班。这次的学习
【培训心得体会】 日期:2022-09-24
-
扶眉战役纪念馆心得体会11篇
扶眉战役纪念馆心得体会11篇扶眉战役纪念馆心得体会篇1有那么一段历史,低诉着血和泪的故事,慢慢地,随岁月老去;有那么一群人,放弃了闲逸的人生,辗转奔波中
【学习心得体会】 日期:2022-08-03
-
凝聚三种力量发展全过程人民民主心得体会12篇
凝聚三种力量发展全过程人民民主心得体会12篇凝聚三种力量发展全过程人民民主心得体会篇1新民主主义革命是指在帝国主义和无产阶级革命时代,殖民地半殖民地国家中的
【学习心得体会】 日期:2022-08-31
-
2022年全国检察长会议心得7篇
2022年全国检察长会议心得7篇2022年全国检察长会议心得篇1眼睛是心灵上的窗户,我们通过眼睛才能看到世间万物,才能看到眼前这美好的一切。拥有一双明亮的眼
【学习心得体会】 日期:2022-10-31
-
全面从严治党的心得体会800字7篇
全面从严治党的心得体会800字7篇全面从严治党的心得体会800字篇1中国特色社会主义是我们党领导
【学习心得体会】 日期:2022-12-14
-
教师全国两会精神学习专题研讨交流材料6篇
教师全国两会精神学习专题研讨交流材料6篇教师全国两会精神学习专题研讨交流材料篇1通过对两会精神深入系统的学习,作为新一代的青年人,更应该严格要求自己,贯彻落
【教师心得体会】 日期:2022-08-11
-
 2024年主题教育民主生活会批评与自我批评意见(38条)(范文推荐)
2024年主题教育民主生活会批评与自我批评意见(38条)(范文推荐)
2023年主题教育民主生活会六个方面个人检视、相互批评意见:1 理论学习系统性不强。学习习近平新时代中国特色社会主义思想不深不透,泛泛而学的时候多,深学细照的时候少,特...
【邓小平理论】 日期:2024-03-19
-
 2024年交流发言:强化思想理论武装,增强奋进力量(完整)
2024年交流发言:强化思想理论武装,增强奋进力量(完整)
习近平总书记指出:“一个民族要走在时代前列,就一刻不能没有理论思维,一刻不能没有思想指引。”党的十八大以来,伴随着新时代中国特色社会主义思想在实践中形成发展的历程...
【三个代表】 日期:2024-03-19
-
2024年度镇年度县乡人大代表述职评议活动总结
xx镇20xx年县乡人大代表述职评议活动总结为响应县级人大常委会关于开展县乡两级人大代表述职评议活动,进一步激发代表履职活力,加强代表与人民群众的联系,提高依法履职水平...
【马克思主义】 日期:2024-03-19
-
 “千万工程”经验学习体会(研讨材料)
“千万工程”经验学习体会(研讨材料)
“千万工程”是总书记在浙江工作时亲自谋划、亲自部署、亲自推动的一项重大决策,也是习近平新时代中国特色社会主义思想在之江大地的生动实践。20年来,“千万工程”先后经历...
【三个代表】 日期:2024-03-19
-
 2024年在市政协机关工作总结会议上讲话
2024年在市政协机关工作总结会议上讲话
同志们:刚才,XX同志对市政协机关20XX年工作进行了很好的总结,很精炼,很到位,可以感受到去年机关工作确实可圈可点。XX同志宣读了表彰决定,机关优秀人员代表、先进集体代...
【邓小平理论】 日期:2024-03-18
-
 在全区防汛防涝动员暨河长制工作推进会上讲话提纲【完整版】
在全区防汛防涝动员暨河长制工作推进会上讲话提纲【完整版】
区长,各位领导,同志们:汛期已经来临,我区城区防涝工作面临强大考验,形势不容乐观。年初,区城区防涝排渍指挥部已经召开专题调度会,修订完善应急预案,建立网格化管理机...
【马克思主义】 日期:2024-03-18
-
 2024年镇作风整治工作实施方案(完整文档)
2024年镇作风整治工作实施方案(完整文档)
XX镇作风整治工作实施方案为深入贯彻落实党的二十大精神及省市区委深化作风建设的最新要求,突出重点推进干部效能提升,坚持不懈推动作风整治工作纵深发展,根据《关于印发《2...
【毛泽东思想】 日期:2024-03-18
-
2024市优化法治化营商环境规范涉企行政执法实施方案【优秀范文】
xx市优化法治化营商环境规范涉企行政执法实施方案为持续优化法治化营商环境,激发市场主体活力和社会创造力,规范行政执法行为,创新行政执法方式,提升行政执法质效,着力解...
【毛泽东思想】 日期:2024-03-18
-
 2024年度关于开展新一轮思想状况摸底排查工作通知(完整)
2024年度关于开展新一轮思想状况摸底排查工作通知(完整)
关于开展新一轮思想状况摸底排查工作的通知为深入贯彻落实关于各地开展干部职工思想状况大摸底大排查情况上的批示要求和改革教育第二次调度会议精神,有针对性做好队伍教育管...
【三个代表】 日期:2024-03-18
-
2024年公路养护中心主任典型事迹材料(完整文档)
“中心的工作就是心中的事业”——公路养护中心主任典型事迹材料**,男,1976年6月出生,1993年参加工作,2000年4月调入**区交通运输局工作,大学本科学历,中共党员,现任**...
【马克思主义】 日期:2024-03-17