氟化物势能函数和热力学性质的分子模拟研究进展
杨松涛,李东洋,牛玉清,李鑫钢,康绍辉,李洪,叶开凯,周志全,高鑫,3
(1 天津大学化工学院,精馏 技术国家工程研究中心,天津 300072;
2 郑州大学化工与能源学院,河南 郑州 450001;
3 物质绿色创造与制造海河实验室,天津 300192;
4 核工业北京化工冶金研究院,北京 101149)
与传统燃煤发电相比,核能发电具有高效、清洁的工业优势,为21 世纪缓解地球温室效应,也为实现我国近年“碳达峰”“碳中和”双碳战略提供了重大机遇[1]。目前我国的核能体系采用的是铀基燃料,铀纯化转化是一个将铀水冶厂所产粗铀浓缩物加工为核纯级六氟化铀(UF6)的过程,是核燃料循环中的至关重要一环。目前世界上铀纯化转化有两条技术路线:湿法与干法。湿法铀纯化工艺技术成熟,建设相对容易,但工艺流程冗长,涉及工序较多,因此投资较大,废液产出也较多,不适应当前绿色发展、清洁生产的先进理念。而干法纯化则充分利用了铀转化工序自身的净化作用,大幅缩短了工艺流程,节省了投资、降低了成本,同时避免了湿法纯化过程中硝酸、磷酸三丁酯(TBP)、煤油和氨等试剂的使用,消除了放射性液体废物的产生,大大减少了放射性废液的产生量和处理量,对确保铀转化的环境安全、改善工作环境具有非常重要的意义。但是,我国对干法铀纯化工艺的研究基本处于空白,特别是干法铀纯化工艺中关键的精馏纯化技术研究还未起步,与国外先进技术差距甚大,这主要归因于氟化物热力学基础数据的大量缺失。在UF6的实际生产过程中,氟化过程会产生大量杂质,比如铬、钼、钨、钒、钛等杂质元素,这对UF6产品质量影响重大。氟化物在化工中也有广泛的应用,如图1 所示,WF6在半导体工业中常用于气相沉积法形成钨膜,SF6常用作变压器中的绝缘气体,CF4是微电子工业中用量最大的等离子蚀刻气体,SiF4常作为生产多晶硅及其衍生物的原料等。此外,图1 还列举了各氟化物的构型与物性。由此可见,获取以上各氟化物的热力学基础数据,尤其是组分间的相平衡数据,对于有效分离工艺的设计至关重要。

图1 氟化物的分子构型、物性及工业应用Fig.1 Molecular configuration,physical properties and industrial applications of fluorides
氟化物体系中杂相样品的获取较为困难,且遇潮湿空气能产生微量HF,使体系具有极强的腐蚀性。同时,精馏纯化的工艺设计涉及高温、高压的操作条件。因此,通过传统实验法获取氟化物体系的热力学性质安全性低、经济性差、操作难度大。分子模拟技术因高经济性、不受苛刻实验条件限制等优势,为解决以上挑战提供了机遇。目前最常见的模拟汽液相平衡的方法是Gibbs Ensemble Monte Carlo(GEMC)方法,该方法已被成功应用于二元或多元汽液、液液相平衡数据的准确计算[2-7]。为实现UF6体系的GEMC 模拟,研究者们已经应用了数种力场模型和独立于温度的势能函数(temperature independent intermolecular potential,TIIP),然而这些模型或是自身拟合精 度 不 足[ 如 Lennard-Jones(12, 6)、Modified Buckingham(exp-6)、Kihara 等][8-14],或是模型复杂度过高(如MMSV、ESMSV 等)[15-17],对模拟效率和准确性有较大影响。为解决以上问题,研究者们提出了与温度相关的势能函数(temperature dependent intermolecular potential,TDIP)[18-23],经比较其模拟准确性得到了极大提升,但仍存在势能函数和参数迁移性问题。
本文对过去数十年分子模拟在氟化物热力学性质模拟计算的进展情况进行了总结分析。首先,分类介绍了汽液相平衡等热力学性质的模拟方法。其次,介绍了不同力场模型,如全原子力场(All Atoms,AA)、联合原子力场(United Atoms,UA)和势能函数(TIIP 和TDIP)的使用范围和发展情况,指明了对氟化物体系的适用性。随后,综述了氟化物热力学性质的分子模拟进展,分析并评价了力场与势能函数选择对计算结果的影响,指出目前存在的问题。最后,对分子模拟方法应用于氟化物热力学性质的计算进行了展望,期待为我国干法铀纯化工艺的设计提供关键的基础数据,推动核工业的发展。
通过以统计热力学为基础的Monte Carlo(MC)模拟能够准确地估计分子性质,从而计算体系p-VT关系、内能、焓、气液相共存(平衡)等宏观性质。典型的宏观性质模拟计算都在特定的统计力学系综中进行,即通过对宏观性质相同但微观性质不同的大数目体系集合的统计平均来实现。如在一个等温、等压和等分子数的系综(NVT)中,某宏观性质A的系综平均通过式(1)获得:

式中,Ai为在量子态i下性质A的数值;
pi为第i个量子态出现的概率;
为系综平均。以下分别介绍氟化物各重要热力学性质的模拟计算原理。
1.1 体系能量

以上各势能项的计算均可通过定义力场来实现,即通过理论计算或实验来确定势能的函数形式和力场参数。在进行UF6、SF6和CF4等氟化物的MC模拟时,通常采用联合原子力场模型[22]来计算体系的总能量,即认为氟化物分子为刚性的正八面体/正四面体结构,不考虑静电势(∑Ecoul)和成键项(∑Evalence),因此其势能仅由成对势(∑Epair)决定。成对势(∑Epair)最广泛采用的函数形式是Lennard-Jones(12,6)势[8-11]:

式中,r为两个分子质心之间的距离,Å(1 Å=0.1 nm);
ε为阱深(well depth),乘以-1对应分子间势能 的 极 小 值(Umin),K;
σ为 碰 撞 直 径(collision diameter),为势能等于零时的分子间距,Å。
因此,体系的焓值也能通过MC 模拟经式(4)而系综平均得到:

在恒温恒容系综(NVT)下,通过粒子数和势能的涨落(即体系总能量的均方差δ)能够模拟出体系的恒容热容[24]:

同样,在恒温恒压系综(NPT)下,体系的恒压热容可以经由式(6)模拟获得[24]:
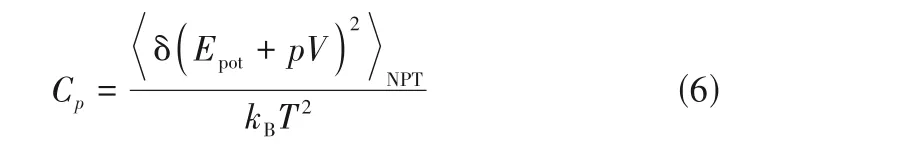
式中,kB为Boltzmann常数。目前,Dove等[25-26]通过Lennard-Jones 势能函数和联合原子力场模型,利用分子动力学(MD)模拟了氟化物的热容等热力学性质,获得了较好的效果。
1.2 p-V-T关系
p-V-T关系常通过状态方程表示,与分子间作用势能紧密相连,能从统计热力学或分子动力学直接推导得出。以维里(Virial)方程为例,形式如式(7)所示:

式中,Z为压缩因子;
V为摩尔体积;
B、C分别为第二、第三维里系数,是物性和温度的函数。第二维里系数(B,second Virial coefficient)反映了二分子的相互作用与分子间势能函数有关。球形分子的第二维里系数计算式如式(8)所示:
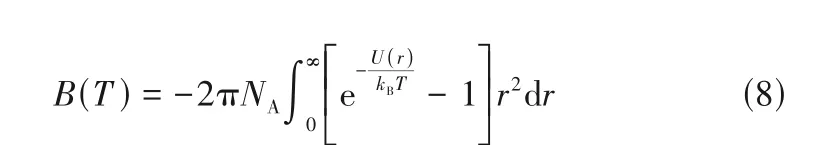
式中,NA为阿伏伽德罗常数;
U(r)为分子间距为r时的分子间势能。Malyshev 等[8-10]利用Lennard-Jones 势能函数和联合原子力场参数模拟计算了氟化物的第二维里系数。
1.3 汽液相平衡
在分子模拟中,MC 法以统计理论为基础,通过随机抽样统计平均得到宏观热力学性质,实现汽液相平衡的模拟计算。根据相平衡状态判断方法的不同,可以将MC 方法分为直接模拟法、间接模拟法以及近几年提出的新方法(表1)[27]。

表1 Monte Carlo法的分类及特点Table 1 Classification and characteristics of Monte Carlo method
直接模拟法将模拟体系划分为气/液或液/液两相主体以及相界面三个区域,当两相的温度、压力及组成不变时即为平衡状态。直接模拟法主要包括GEMC 方法[2-7],适合简单流体和较复杂流体相平衡分子模拟预测;
构型偏倚的Monte Carlo 方法(configuration bias Monte Carlo,CBMC)[28],适合复杂大分子或含有氢键等强相互作用的稠密流体的相平衡分子模拟预测;
应用反应系综的Monte Carlo 模拟法(reaction Gibbs ensemble Monte Carlo,RGEMC)[29],适合模拟反应体系的相平衡。
间接模拟法通过一系列化学势计算来判断模拟是否达到平衡状态。在一定的温度压力下,当气/液两相的化学势相等时即为平衡状态。间接模拟法主要包括NPT+测试粒子法(NPT+TP,NPT + test particle)[30],模拟时间长且需要通过标准热力学关系对化学势进行校正,过程较为烦琐,因此应用较少;
巨正则系综Monte Carlo 模拟方法(Gibbs canonical Monte Carlo,GCMC)[31],适合非均相系统的模拟研究;
Gibbs-Duhem 积分法(Gibbs-Duhem integration,GDI)[32],主要用于纯物质的计算,应用较多;
直方图再加权法(histogram reweighting,HRW)[33],主要应用于气液相波动剧烈的临界区的模拟。
为了提高相平衡模拟的准确性和有效性,近几年研究者们在上述算法的基础上加以改进,提出了新的模拟方法。如HRW 和GEMC 耦合的方法[34],其计算结果比GEMC 方法更加准确,但尚未应用到混合物的模拟中;
动力学Monte Carlo 模拟方法(kinetic Monte Carlo,kMC)[35-36],适合模拟稀薄流体和缔合流体的相平衡;
Bin-CMC(Bin canonical Monte Carlo)模拟方法[37-38],不仅可以模拟汽液相平衡,还可以模拟气固相平衡。
考虑到UF6等氟化物的分子结构(图1)均为非极性的球对称型分子(四面体或八面体构型),因此首选GEMC方法模拟其相平衡热力学性质[22](图2)。模拟流程简介如下:首先,将氟化物分子视为假原子,使用TIIP 或TDIP 势能函数描述分子间相互作用。然后,建立相对独立但热力学相关的两个模拟盒子,分别代表气液两相主体。当模拟纯组分的气液共存性质时,通常规定两盒的温度、总分子数及总体积不变(即NVT 系综);
当模拟二元或多元混合物的汽液相平衡性质时,则需要规定两个盒子的温度、压力及总分子数不变(NPT系综)。通过一系列Monte Carlo运动使两盒达到汽液相共存或相平衡状态。最后,由系综平均计算气、液两相的热力学性质。

图2 氟化物相平衡热力学性质的GEMC方法Fig.2 GEMC method for thermodynamic properties of fluoride phase equilibrium
分子力场是描述分子内以及分子间相互作用的势能函数。根据对分子的抽象程度不同,可以将力场分为全原子力场(all-atom force field,AA)、联合原子力场(united-atom force field,UA)和粗粒化力场(coarse-grained force field,CG)等。全原子力场(AA)考虑分子中的所有原子,通常涉及的温度压力范围较小,计算比较耗时,但结果较为精确,如OPLS-AA[39]、COMPASS[40]和CHARMM[41]等。联合原子力场(UA)将原子基团(如甲基、亚甲基)作为一个相互作用位点,通常具有更大的温度压力范围和更高的计算效率,对相平衡热力学性质的计算较为精确,特别适合于包含粒子交换等Monte Carlo 运动的GEMC 模拟计算,但对其他性质(如黏度)的预测存在一定的误差,如TraPPE-UA[42-44]和NERD[45]等。粗粒化力场则进一步抽象分子结构,将更大的原子团(有时称为珠子、颗粒或超原子等)作为一个相互作用位点,计算效率更高,主要用于蛋白质等生物大分子,如Martini[46]和SDK[47]等。由于UF6、SF6和CF4等氟化物的分子为简单的球对称型分子,因此选用联合原子力场(UA)能在保证对物性准确描述下实现汽液相平衡的高效模拟。值得注意的是,Olivet 等[48]提出了SF6的全原子力场,并通过分子动力学(MD)模拟了SF6的相平衡性质和传递性质,获得了比联合原子力场更精确的预测效果。
在力场中,根据原子序数和所处的化学环境将同种元素划分为不同的原子类型(atom type),每种原子类型对应各自的力场参数。力场开发的过程就是力场中相互作用势能函数的确定以及力场参数化的过程[27]。早期的力场参数大多通过拟合实验数据得到,随着计算机及量子力学技术的发展,从头算(ab initial)成为力场参数化的主流方法,但该法在处理范德华相互作用时存在较大困难,因此范德华相互作用参数仍通过拟合第二维里系数、液体密度和蒸发焓等得到。为拟合范德华相互作用参数,研究者们还开发了多种分子间势能函数模型。简单的球对称势能模型(如硬球模型、软球模型和方阱势能模型)只适用于惰性气体和轻球形分子(如甲烷)[49]。对于UF6、SF6和CF4等氟化物分子,多数研究者使用各向同性分子间势(isotropic intermolecular potential)来描述其范德华相互作用,如Lennard-Jones(12,6)、Modified Buckingham(exp-6)和Kihara 等[8-17]。这些势能的特点是分子间势能参数独立于温度(TIIP),即碰撞直径(σ)和阱深(ε)不随温度变化。
2.1 独立于温度的分子间势能函数
2.1.1 Lennard-Jones(12,6)势 Lennard-Jones(12,6)势常用来描述非极性分子,对单原子分子(如惰性气体)和简单的球形分子尤其有效。该势能函数形式简单、计算效率高、使用最为广泛。UF6、SF6和CF4等氟化物分子为八面体或四面体对称结构,偶极矩约等于零,适用于Lennard-Jones(12,6)势,其计算形式如式(3)所示。
将排斥指数n(replusion parameter)作为可调参数,可以控制泡利(Pauli)排斥作用的大小,得到更具一般性的Lennard-Jones(n,6)势能函数,如式(9)所示:

图3展示了排斥指数(n)分别为9、12和18时的Lennard-Jones(n,6)势能曲线。随着n的增加,吸引势阱的宽度变窄,陡度增大。

图3 Lennard-Jones(n,6)势能曲线Fig.3 Lennard-Jones(n,6)potential curves
2.1.2 Modified Buckingham(exp-6)势 理论研究表明,分子间斥力随分子间距离的增大呈指数衰减(即U- e-ar)[50],因此Lennard-Jones(12,6)的12次项无法真实表现分子间的排斥相互作用。在此基础上,Rice 等[51]提出了Modified Buckingham(exp-6)势能,如式(10)所示:

式中,rm为平衡距离(equilibrium distance),Å,此时分子间势能最小(Umin);
参数a衡量了排斥能(repulsive potential)曲线的陡峭程度。图4 展示了Modified Buckingham(exp-6)势能曲线。

图4 Modified Buckingham(exp-6)势能曲线Fig.4 Modified Buckingham(exp-6)potential curves
2.1.3 Kihara 势 Kihara[52]考虑了分子本身的大小对分子间势能的影响,提出了Lennard-Jones(12,6)的改进形式,即Kihara势,如式(11)所示:

式中,a为硬核半径(hard core radius),Å。
2.1.4 MMSV 势 无 论 是Lennard-Jones(12,6)势,还是经过改进的exp-6 和Kihara 势,均高估了分子间的排斥作用,无法精确预测氟化物的热力学 性 质[15]。因 此,Aziz 等[15]在Parson 等[53-55]提 出 的分段势的基础上,构造了一种新的MMSV(Morse-Morse spline van der Waals)势,如式(12)和式(13)所示。

式中,x=r/rm。由势能曲线的连续性条件及其斜率确定样条曲线参数(a1、a2、a3、a4);
通过从头算确定色散系数(C6、C8、C10);
通过拟合黏度、热导率、维里系数等实验数据确定剩余6 个参数(ε、rm、x1、x2、β1、β2)。
2.1.5 ESMSV势 为了能够同时精准地预测UF6的第二维里系数、黏度和碰撞诱导光散射光谱(collision-induced light scattering,CILS),El-Kader等[17]构造了另一种分段势,即ESMSV (exponential spline morse spline van der Waals) 势,如 式(14)和 式(15)所示:

由势能曲线的连续性条件及其斜率确定样条曲线参数(A1、A2、A3、A4、B1、B2、B3、B4);
通过从头算确定色散系数(C6、C8、C10),通过拟合黏度、热导率、维里系数等实验数据确定剩余9个参数(ε、rm、A0、B0、ξ、r1、r2、r3、r4)。
然而,研究发现UF6、SF6和CF4等氟化物分子的热力学性质对温度的敏感性较强[18-23],因此上述势能模型在预测其第二维里系数等热力学性质时,存在拟合精度不足或模型复杂度过高等问题,对模拟效率和准确性有较大影响[8-17]。例如,Lennard-Jones模型的计算结果常常表现为高温时产生负偏差,低温时产生正偏差[56]。其原因可能有以下两点:一是维里展开理论不适用于低温下的稠密气体和液体;
二是对于较大温度范围内的重分子或复杂分子,不应将势能参数作为常数[57]。Meinander 等[58]在研究SF6的CISL 光谱数据时发现,氟化物分子间相互作用的各向异性会使其分子间势能略微依赖于温度,即碰撞直径(σ)随温度的升高略有增加,阱深(ε)随温度的升高而减小。
2.2 与温度相关的分子间势能函数
在Meinander 等[58]的研究基础上,Stefanov 等[59]考虑了振动激发对UF6、SF6和CF4等重球形分子(heavy globolar molecular,HGM)间相互作用的影响,提出了分子振动激发态模型(vibrationally excited states of molecules,VESM)。将重球形分子气体作为若干个振动激发态的混合物,振动激发会导致分子有效尺寸的增大,且分子量越大,分子的振动激发效应越大[60]。利用VESM 模型,Stefanov 等[61]得到了SF6的半经验分子间相互作用势能参数。结果显示,碰撞直径(σ)随温度升高而增加,阱深(ε)随温度升高而减小,这与Meinander 等[58]的结论相同。值得注意的是,振动激发态数ns会随分子量的增加急剧增大,而VESM 模型需要计算的势能数与振动激发态数ns成正比[等于ns(ns+1)/2],因此该模型的计算过程相当复杂且耗时。例如对于UF6,使振动配分函数收敛需要200000步左右[18]。
近年来,Zarkova 等[20-21]通过引入与温度相关的分子间势能函数(TDIP),克服了VESM 模型的上述缺点。该模型适用于球形分子,以及至少包含一种球形分子的二元混合物。其改进之处在于,用一个平均态来代替VESM 模型中在给定温度T下的ns个振动激发态,则平均分子增大半径δ(eff)可以用式(16)来计算:

TDIP模型将VESM模型中的ns个振动激发态的混合物作为一种具有与温度相关的平均有效尺寸的等分子气体。因此,在给定温度T下,可以用各向同性分子间势来描述其相互作用。如果选择Lennard-Jones(n,6),则势能函数U(r,T)的计算形式如式(17)~式(19)所示:

式中,rij为两个分子质心间的距离,Å;
εij为阱深,K;
σij为碰撞直径,Å;
δ为温度放大系数。
以上是温度相关函数的双曲线形式。此外,还有线性函数形式[62]和双指数函数形式[63]。
在分子模拟中,第二维里系数的计算较为简单直接[式(8)],且通过第二维里系数能够间接计算物质的气液共存数据。因此,研究者们通常首先对第二维里系数的实验值和计算值进行比较,量化势函数模型的精度。
3.1 TIIP应用于氟化物热力学性质模拟
Lennard-Jones(12,6)势形式简单、计算效率高、使用最为广泛,是描述氟化物分子间相互作用的首选。Malyshev[8-9]对其测量的第二维里系数进行拟合,得到了UF6、MoF6和WF6等的Lennard-Jones(12,6)势能参数,其结果只对低温下的第二维里系数的预测效果相对较好,无法预测氟化物的传递性质(黏度η、扩散系数D等)。此后,Morizot 等[10]从其测量的第二维里系数和黏度拟合出了新的Lennard-Jones(12,6)势能参数,虽然其拟合参数适用的温度范围较窄,但在预测混合物(UF6-MoF6和UF6-WF6)的黏度和第二维里系数方面表现较好。1974 年,Schneider 等[11]利用量子力学方法得到了UF6的Lennard-Jones(12,6)理论计算参数,但是不能预测Heintz 等[12-13]测量的第二维里系数和传递系数(黏度η、扩散系数D等)。2016 年,Al-Matar 等[22]对Dymond 等[64]提出的第二维里系数关联式进行拟合,得到了UF6的Lennard-Jones(12,6)势能参数,其结果很好预测了UF6的第二维里系数,均方根偏差(root mean square deviation,RMSD)为11.813。Al-Matar 等[22]还利用GEMC 方法模拟了UF6的气液共存曲线,但结果显示与UF6的实验密度数据误差较大[65-66]。
由于Lennard-Jones(12,6)势未考虑分子本身大小对分子间力的影响,且在描述分子间斥力时存在理论缺陷,因此其他研究者使用了Modified Buckingham(exp-6)势、Kihara 势和Lennard-Jones(n,6)势来描述氟化物的分子间相互作用。1965 年,Kirch 等[14]对其测量的UF6自扩散系数进行拟合,得到了UF6的Modified Buckingham(exp-6)势能参数,结果表明该模型比Lennard-Jones(12,6)的计算难度大,且无法准确预测UF6的传递系数和第二维里系数。1974 年,Malyshev[67]对其测得的第二维里系数进行拟合,得到了UF6的Kihara 势能参数,但该模型不能同时兼顾UF6的第二维里系数和传递系数(黏度η、扩散系数D等)的准确计算。1976年,Heintz等[12-13]测量了一些准球对称分子(UF6、MoF6和WF6等)的第二维里系数(B)和传递系数(黏度η、扩散系数D),但值得注意的是,其第二维里系数的测量值与Malyshev[8-9]和Morizot 等[10]的相差1.4 倍。Heintz等[12-13]对其测量值进行拟合,得到了UF6、MoF6和WF6等的Lennard-Jones(n,6)势能参数,该势能模型增加了排斥指数作为第三个参数(n=13.58),其预测的第二维里系数系统性高于其他研究者的测量值。
为了能够同时精确预测氟化物的黏度和第二维里系数,研究者们提出了分段势模型[53-55],结果显示可以更好地预测氟化物的热力学性质。1991 年,Aziz 等[15]利用MMSV 势能模型精准预测了SF6的第二维里系数和黏度数据。该势能所预测的黏度与多位研究者所测的大部分实验数据误差都在1%以内,所预测的维里系数在307 K 以上与实验值几乎完全一致。在此基础上,Coroiu 等[16]计算了描述球形分子(特别是六氟化物)的简化第二维里系数(reduced second virial coefficients)、Enskog-Chapman碰撞积分和传递系数的更高近似值,确定了UF6的MMSV 势能参数。其计算结果与实验值的偏差为0.2%~4.5%(黏度)、1.1%~4.1%(自扩散系数)和0.1%~3.5%(第二维里系数)。这表明,MMSV 势能模型对六氟化物的相平衡性质和传递性质的预测效果均优于之前所报道的势能模型。然而,计算过程复杂以及缺乏其他氟化物分子精确的C6色散系数等原因限制了其应用范围。2016 年,El-Kader 等[17]对UF6的第二维里系数、黏度和碰撞诱导光散射光谱(CILS)实验数据进行拟合,得到了UF6的ESMSV 势能参数。虽然该模型对上述实验数据的预测能力较好(RMSD=0.93),但是其计算过程比MMSV 更复杂(涉及11个参数)。
综上所述,TIIP 模型在预测氟化物相平衡性质(第二维里系数B)和传递性质(黏度η、自扩散系数D)时,存在自身拟合精度不足[如Lennard-Jones(12, 6),exp-6 和Kihara 等]以及模型复杂度过高(如MMSV 和ESMSV 等)等问题,对模拟效率和准确性有极大影响。
3.2 TDIP应用于氟化物热力学性质模拟
Zarkova 等[20]采用Lennard-Jones(n,6)势能模型并使用了温度相关函数(TDIP),先后对BF3、CF4、SiF4、CCl4、SiCl4、SF6、MoF6、WF6、UF6、C(CH3)4和Si(CH3)4共11 种重球形分子气体的第二维里系数(B)、黏度(η)和自扩散系数(D)进行拟合,得到了平衡距离(rm)和阱深(ε)作为温度的幂的多项式。图5展示了UF6的rm(T)和ε(T)的温度依赖关系。在200~800 K 的温度范围内,平衡距离(rm)变化了约10%,而阱深(ε)变化了约40%。该图表明,UF6较大的分子量使其温度效应非常剧烈,在进行较大温度范围内UF6的分子模拟计算时,不能将rm和ε作为常数。

图5 UF6的rm(T)和ε(T)的温度依赖关系Fig.5 Temperature dependence of rm(T)and ε(T)of UF6
Kestin 等[68-75]以SF6(实验和理论上研究最多的重球形分子)为例,将利用TDIP 方法获得的计算黏度数据与精度至少为0.5%的实验黏度数据进行比较, 绝对偏差在1%以内;
再与其他研究者[75-76]的计算黏度数据比较,绝对偏差为:<1%(Boushehri)和<0.4%(Trengove & Wakeham)。将利用TDIP 方法获得的计算第二维里系数数据与Boushehri等[76-77]的计算数据比较,偏差在±6 cm3/mol以内。
同时,Zarkova 等[20]还绘制了SF6的TDIP 势能曲线(300 K 和600 K),并与精度较高的MMSV 势能进行比较(图6)。结果显示,MMSV 具有较小的碰撞直径(σ)和较大的阱深(ε),但是Lichtenthaler 等[78]的晶体学数据(σ=5.02310210 Å)更接近TDIP 的参数值(5.04310210 Å)。此外,TDIP 方法不需要C6色散系数的测定,且已成功应用于不同的HGM分子及其混合物[18-21],而MMSV 势能目前的研究成果仅限于纯物质(SF6和UF6等)[15-16]。

图6 TDIP与MMSV势能曲线的比较(SF6)[15]Fig.6 Comparison of potential curves between TDIP andMMSV(SF6)[15]
Zarkova 等[21]进一步将TDIP 方法推广到二元混合物,确定了102 种重球形气体二元混合物在200~900 K 温度范围内的分子间相互作用势能参数(包含CH4-CF4、SF6-CF4、UF6-MoF6、UF6-WF6和MoF6-WF6等),精准预测了其传递性质(混合物黏度ηmix)和相平衡性质(混合物第二维里系数Bmix)。例如,对于CH4-CF4和SF6-CF4等二元体系,B12(二元维里交互作用系数)的偏差比Bzowski 等[79]报道的要小;
对于CH4-CF4、SF6-CF4和CH4-SF6等二元体系,ηmix和Bmix的计算值与实验数据也具有良好的一致性;
对于UF6-MoF6、UF6-WF6和MoF6-WF6等二元体系,ηmix计算值与实验值的偏差小于1%。研究结果证实了TDIP的可靠性和普遍性。此外,Zarkova等[80]还比较了Lorentz-Berthelot (LB)和Tang-Tonenies (TT)混合规则对拟合结果的影响:在计算相平衡性质时,LB优于TT;
而在计算传递性质时,TT略优于LB。
在Zarkova 等[18]的研究基础上,Al-Matar 等[22]进一步利用GEMC 技术模拟了UF6的气液共存性质。其利用修改和简化的温度相关函数[式(20)~式(22)]对第二维里系数实验数据进行拟合,得到了与Zarkova 等不同的TDIP 参数集(图7):二者的碰撞直径(σ)接近,且均与温度成正比(在300~480 K 的温度范围内均增大了约3%),但阱深(ε)相差很大(Al-Matar 等为323.63 K,Zarkova 等为1044 K)。原因是二者所采用的排斥指数(n)不同(Al-Matar等为12,Zarkova等为27.81)。
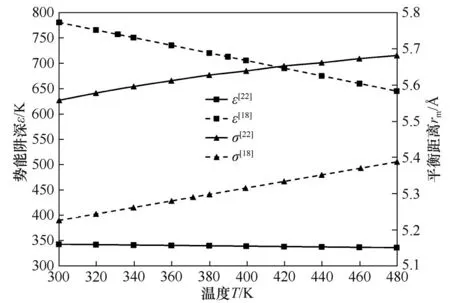
图7 温度对UF6碰撞直径(σ)和阱深(ε)的影响Fig.7 Effect of temperature on UF6 collision diameter(σ)and well depth(ε)
Al-Matar 等[22]以Dymond 等[64]提供的UF6第二维里系数关联式[式(23)]为基准,计算了不同文献中UF6的第二维里系数理论计算值的均方根偏差(RMSD)[式(24)],结果如表2所示。Al-Matar等使用TIIP 法计算得到的第二维里系数的RMSD 为11.187,而TDIP 法 的 为3.8931,比TIIP 减 少 了 约60%。
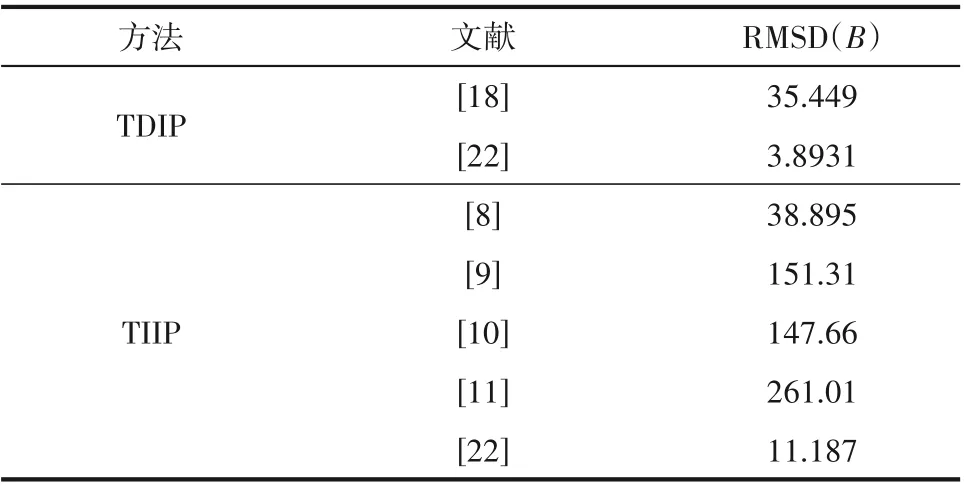
表2 不同文献中UF6的第二维里系数的RMSD Table 2 RMSD of second virial coefficient of UF6 in different literatures

式中,N为观测次数;
Ai,exp、Ai,cal分别为第i次观测的实验值和理论计算值。
Al-Matar 等[22]利用其拟合得到的TIIP 参数和TDIP 参数分别模拟了UF6的气液共存曲线(图8)。结果显示,TDIP 方法提高了气液共存曲线的模拟精度,但是和UF6的实验密度数据仍有一定差距[65-66]。他们指出TDIP 使精度提高的原因可能是降低了阱深(ε),从而降低了分子间吸引力的大小,而精度仍然不高的原因可能是:(1)UF6的联合原子方法(UA)的不足;
(2)Lennard-Jones模型本身的不足;
(3)未考虑电荷间静电相互作用的影响。Al-Matar等的研究结果表明,TDIP 在UF6热力学性质的模拟方面具有明显优势,与传统的TIIP相比,在第二维里系数和气液共存性质等的模拟计算中具有较高的准确性。

图8 UF6的气液共存模拟曲线(TDIP和TIIP)与实验数据Fig.8 Vapor-liquid coexistence curves(TDIP and TIIP)and experimental data of UF6
Oh[23]在 拟 合SF6、MoF6、WF6、UF6、C(CH3)4和Si(CH3)4等的Kihara 势能参数时,也使用了与温度相关的势能函数。将基团贡献概念应用到Kihara 势中,即六氟化物分子作为一个单分子基团,而将四甲基分子的官能团指定为CH0、CH3、Si0。为了使模型得到简化,增加模型的通用性,Oh 采用球心模型(spherical core model),并将阱深(ε)作为与温度相关的势能参数,如式(25)所示。通过对第二维里系数、黏度等实验数据进行拟合,得到了上述分子基团和官能团的Kihara 势能参数,计算了与实验值的均方根偏差(RMSD)。表3 以UF6为例,总结了第二维里系数和黏度的计算关联式与实验值的RMSD。从表中可以看出,与Tsonopoulos[81]、Zarkova 等[20]的关联式相比,基团贡献法能更好地预测上述重球形分子的第二维里系数,但略差于Dymond等[64]的关联式;
基团贡献法的黏度计算精度高于Lucas[82]的关联式,但低于Zarkova等[20]的关联式。此外,Oh还计算了26个基团的二元相互作用系数,成功预测了混合物性质[23]。


表3 UF6第二维里系数和黏度的计算关联式与实验值的RMSDTable 3 RMSD of calculation correlation and experimental data of second virial coefficient and viscosity of UF6
Zarkova 等、Al-Matar 等以及Oh 等的研究成果,证明了TDIP 方法在分子模拟热力学性质领域中的应用潜力。此外,其他研究者将TDIP方法的应用范围拓展到双中心LJ分子[57],进一步增强了TDIP方法的实用性。
本文对有关氟化物热力学性质的分子模拟研究进展进行了综述。UF6、SF6和CF4等氟化物是简单的球对称型分子,然而受振动激发效应的影响,这类分子的势能参数是与温度相关的函数(TDIP),即分子间的碰撞直径(σ)随温度的升高而增加,阱深(ε)随温度的升高而减小。已有的研究结果表明,TDIP 在上述体系中具有明显优势,与传统的独立于温度的分子间势能参数(TIIP)相比,在第二维里系数、黏度、气液共存性质等的模拟计算中具有较高的效率和准确性,但要获取精确的氟化物热力学数据,尚有很多基础科学问题需要解决。面临的挑战有以下几点。
(1)由于氟化物实验数据的缺乏且各数据组之间存在较大的偏差,拟合得到的分子间势能参数误差较大,需要完善第二维里系数、密度和蒸发焓等实验数据,对势能参数(σ和ε)和排斥指数(n)进一步优化。
(2)氟化物的热力学性质模拟计算中使用的多为联合原子力场,需要借助量子化学计算优化氟化物的分子结构,获取氟化物的键能参数并探究其对模拟计算的影响,进一步提高氟化物热力学性质模拟计算的效率和准确性。
(3)应用TDIP计算两相混合物的汽液相平衡性质还未取得任何进展,这与缺乏相关实验数据对比有关。因此,需要通过设计安全可靠的汽液相平衡实验来获取更多的混合物实验数据,同时进一步探究不同势能模型和混合规则对模拟计算的影响。
(4)为适应不同工艺设计条件需要,通常需要配套多个T、P下的相平衡数据,因此保证TDIP 模型在较广温度、压力范围内良好迁移性就显得极为重要。这要求进一步优化TDIP 模型参数(rm、ε和δ),以及考察不同TDIP 形式的影响,如线性函数形式、双曲线函数形式和双指数函数形式等。
氟化物体系具有强的化学腐蚀性,利用传统实验法较难获取其相平衡热力学性质,而分子模拟技术环保经济、不受苛刻实验条件的影响,为预测氟化物的相平衡热力学性质提供了机遇,以期为具体工艺设计提供实践基础。然而,国内关于氟化物热力学性质的分子模拟研究尚处于空白阶段,因此,在今后的研究中,获取氟化物的基础热力学实验数据,优化氟化物分子的结构和力场参数,提高氟化物力场模型在不同温度和压力范围内的迁移性,从而更好地描述氟化物的相平衡热力学性质,是发展我国干法铀纯化转化技术的重中之重。
猜你喜欢 氟化物热力学势能 了解固体和液体特性 掌握热力学定律内容中学生数理化(高中版.高考理化)(2022年5期)2022-06-01航空航天类“工程热力学”课程思政建设的探索与实践大学·课外阅读(2022年3期)2022-04-25热力学第一定律易混易错剖析中学生数理化(高中版.高考理化)(2021年5期)2021-07-16大阪地下水有机氟化物超标110倍环球时报(2021-06-28)2021-06-28势能的正负取值及零势能面选择问题初探高中生学习·高三版(2017年9期)2017-10-26“动能和势能”“机械能及其转化”练习中学生数理化·八年级物理人教版(2016年5期)2016-08-26国内主要生产厂砖茶中氟化物含量调查与分析科教导刊·电子版(2016年19期)2016-08-19弹性势能纵横谈新高考·高一物理(2015年3期)2015-08-20关于重力势能和弹性势能理解与运用的几个典型错误新高考·高一物理(2014年4期)2014-09-17促进物理思维训练的好题中学生数理化·高二版(2008年10期)2008-06-17- 范文大全
- 说说大全
- 学习资料
- 语录
- 生肖
- 解梦
- 十二星座
-
主题党日活动交流发言8篇
主题党日活动交流发言8篇主题党日活动交流发言篇13月13日,东城区党史学习教育动员大会召开。市委
【活动总结】 日期:2022-12-23
-
2022年4月主题党日活动记录范文15篇
2022年4月主题党日活动记录范文15篇2022年4月主题党日活动记录范文篇1一个崇尚阅读的民族,必然精神饱满、意气风发、活力四射。习近平总书记强调:“学习
【活动总结】 日期:2022-08-01
-
家乡赋|最美的家乡赋
家乡赋 孙传志 今安康市,白河双丰镇,吾之家乡也。三环沃土,山水环抱。其北依山,山系五岭,山
【调研报告】 日期:2020-04-01
-
【人教版1-6年级数学上册知识点精编】1-6年级数学人教版教材
人教版二年级数学上册知识点汇总第一单元长度单位一、米和厘米1、测量物体的长度时,要用统一的标准去测量
【调研报告】 日期:2020-11-08
-
党支部1-12月全年主题党日活动计划表
2022年党支部主题党日活动计划表序号活动时间活动方式活动内容12022年1月专题学习研讨集中观看2022年新年贺词,积极开展学习研讨交流。组织生活会组织党员认真对照党章...
【活动总结】 日期:2022-10-14
-
2022年2月份主题党日活动记录5篇
2022年2月份主题党日活动记录5篇2022年2月份主题党日活动记录篇1尊敬的党组织:在今年的开学初,本人积极参加教研室组织的教研活动,在学校教研员的指
【活动总结】 日期:2022-08-12
-
2023年平安校园建设方案13篇
平安校园建设方案“平安校园”创建工作,我们幼儿园全体教职员工一直把它当作头等大事来抓。领导高度重视,以“平安校园”创建活动为抓手,建立和规范校园安全工作机制
【规章制度】 日期:2023-11-02
-
医院最佳主题党日活动11篇
医院最佳主题党日活动11篇医院最佳主题党日活动篇1 医院最佳主题党日活动篇2为隆重纪念中国共产党成立100周年,进一步巩固党的群众路线教育实践活动成果,切实
【活动总结】 日期:2022-10-29
-
主题党日活动记录202210篇
主题党日活动记录202210篇主题党日活动记录2022篇12021年是中国共产党成立100周年,为广泛开展爱国主义宣传教育,铭记党的历史,讴歌党的光辉历程,
【活动总结】 日期:2022-08-02
-
南京大屠杀国家公祭日悼念文案句子11篇
南京大屠杀国家公祭日悼念文案精选句子1、惟有民魂是值得宝贵的,惟有他发扬起来,中国才有真进步。——鲁迅2、我爱我的祖国,爱我的人民,离开了它,离开了他们,我
【企划文案】 日期:2023-10-20
-
正式的晚宴邀请函 公司晚宴邀请函
尊敬的先生 女士: 我公司谨定于xxxx年xx月xx日xx:xx在xxxx店隆重举行xx市xx届xxxx晚宴(宴会地址:xx区xx路xxxx) 敬请届时光临!xxxxxx集团股份有限公司xxxx有限公司敬邀xxxx年xx月xx日
【简历资料】 日期:2019-08-03
-
《国行公祭,为佑世界和平》课文原文阅读_国行公祭为佑世界和平每段段意
国行公祭,为佑世界和平钟声“国行公祭,法立典章。铸兹宝鼎,祀我国殇。”侵华日军南京大屠杀遇难同胞纪念
【简历资料】 日期:2020-11-28
-
一年级新学期目标简短_一年级学生新学期打算
新学期到了,我是一年级下册的小学生了。 上课的时候,我要认真学习,不做小动作,认真听讲。我要认真学习,天天向上,努力学习,耳朵要听老师讲课,眼睛要瞪得大大的看老...
【简历资料】 日期:2019-10-26
-
[信访复查复核制度作用探讨]信访复查复核有用吗
作为我国特有的一项制度,信访制度的出现并长期存在不是偶然的,虽然一些法学专家认为信访制度具有“人治”
【职场指南】 日期:2020-02-16
-
[党员干部2019年主题教育个人问题检视清单及整改措施2篇] 党员干部
2019年主题教育问题检视清单及整改措施根据主题教育领导小组办公室《关于认真做好主题教育检视问题整改
【求职简历】 日期:2019-11-08
-
红旗颂朗诵稿原文【《红旗颂》朗诵词】
《红旗颂》朗诵词 女:晴空万里,红旗飘扬, 六十载风云,我们昂首阔步。 男:六十个春秋,
【职场指南】 日期:2020-02-16
-
网络维护工作内容_(精华)国家开放大学电大专科《网络系统管理与维护》形考任务1答案
国家开放大学电大专科《网络系统管理与维护》形考任务1答案形考任务1理解上网行为管理软件的功能【实训目
【职场指南】 日期:2020-07-17
-
党委会与局长办公会的区别_局长办公会制度
为进一步加强xxx局工作的规范化、制度化建设,提高行政效能,规范议事程序,特制定本制度。一、会议形式1、局长办公会议由局长、副局长参加。由局长召集和主持。根据工作需要...
【求职简历】 日期:2019-07-30
-
如何凝心聚力谋发展【坚定信心谋发展凝心聚力促跨越】
当前,清河正处于在苏北实现赶超跨越基础上全面腾飞的战略机遇期,处于在全市率先实现全面小康基础上率先实
【简历资料】 日期:2020-03-17
-
《铁拳砸碎“黑警伞”》警示教育片观后感
影片深刻剖析了广西北海市公安局海西派出所原所长张枭杰蜕变堕落的轨迹。观看警示教育片后,做为一名党员教
【简历资料】 日期:2020-08-17
-
妇女节活动实施方案14篇
妇女节活动实施方案14篇妇女节活动实施方案篇1一、活动背景:为了活跃校园文化氛围,迎接国际妇
【其他范文】 日期:2022-12-24
-
与小朋友互动的暑期社会实践报告:社会实践报告
又到月末了想本月没什么事情,倒是一件事情蛮有意义的,于是著文以记之。 7月16号,进行一次为期一天的暑期社会实践,内容是与小朋友互动,参观宜兴陶瓷博物馆还有亲手...
【口号大全】 日期:2019-07-04
-
论《河洛精蕴》的“数理”变占说
杨易辰江永(1681—1762)字慎修,清代乾嘉时期著名的易学家。他撰写的《河洛精蕴》一书,以“河图
【其他范文】 日期:2023-01-23
-
公司领导庆祝三八妇女节大会讲话稿
女职工同志们:你们好!春回大地,万物复苏。今天,我们在这里隆重集会,纪念“三八”国际劳动妇女节周年。
【其他范文】 日期:2021-05-21
-
银行更改函(精选文档)
当前位置:>>>2021-12-12银行更改函1中国建设银行民族支行:我单位在贵行开立的一般存款帐户(户名为:汽车用品有限公司,账号为:45xx16x4361x8x9xx5x1),于x8年8月18日通
【其他范文】 日期:2022-11-25
-
银行领导班子成员党史学习教育专题民主生活会个人对照检查材料3篇
银行领导班子成员党史学习教育专题民主生活会个人对照检查材料3篇银行领导班子成员党史学习教育专题民主生
【其他范文】 日期:2022-12-25
-
在公司“牢记嘱托 再立新功 再创佳绩 喜迎二十大”主题行动启动会上的讲话
下面是小编为大家整理的在公司“牢记嘱托 再立新功 再创佳绩 喜迎二十大”主题
【其他范文】 日期:2022-10-19
-
空压机维护维修的问题分析与应对
李树军,刘喜,杨勇(1 渤海装备巨龙钢管有限公司,河北沧州062552;2 河北华油一机图博涂层有限
【其他范文】 日期:2023-01-12
-
电访员如何巧用方言订货|访烟订货新商盟官网
电访员在工作中使用普通话与客户交流,是烟草行业自身规范化、标准化、现代化的需要。但电访员所服务的对象
【口号大全】 日期:2020-02-21
-
2022年在退役军人事务局党员冬训动员部署会议上的讲话
在退役军人事务局党员冬训动员部署会议上的讲话同志们,今天我们在这里召开退役军人事务局20xx年度党员冬训工作动员部署会。会议的主题十分明确,就是要深入学习贯彻******新...
【其他范文】 日期:2022-09-28
-
理论中心组学习总体国家安全观发言材料9篇
理论中心组学习总体国家安全观发言材料9篇理论中心组学习总体国家安全观发言材料篇1(八)深入学习贯彻中央以及省的重要会议和文件精神深入学习贯彻年度内中央以
【发言稿】 日期:2022-08-04
-
军转座谈会交流发言4篇
军转座谈会交流发言4篇军转座谈会交流发言篇1大家好,我叫贺丽,2015届选调生,来自康定市委组织部,现在省委编办跟班学习。今天,非常荣幸向大家汇报我的学习收
【发言稿】 日期:2022-10-27
-
12岁生日小寿星发言4篇
12岁生日小寿星发言4篇12岁生日小寿星发言篇1各位来宾、各位朋友:大家好!今天,我们欢聚在这里,共同庆祝**十二周岁生日。首先,我代表**的父母以
【发言稿】 日期:2022-07-31
-
党内警告处分表态发言14篇
党内警告处分表态发言14篇党内警告处分表态发言篇1尊敬的各位领导、同事们:大家上午好!刚才会上宣布了党委关于我任职的决定,我首先衷心感谢党委的信任和
【发言稿】 日期:2022-09-13
-
党内警告处分党员讨论发言3篇
党内警告处分党员讨论发言3篇党内警告处分党员讨论发言篇1大家好!作为新时期的一名大学生,认真学习、深刻领会、全面贯彻省党代会精神,是当前和今后一个时期重
【发言稿】 日期:2022-08-07
-
廉政大会总结发言稿7篇
廉政大会总结发言稿7篇廉政大会总结发言稿篇1各位领导,同志们:根据会议安排,我就党风廉政建设工作做表态发言,不妥之处,请批评指正。一、提高认识,切实
【发言稿】 日期:2022-10-30
-
被约谈的表态发言8篇
被约谈的表态发言8篇被约谈的表态发言篇1各位领导、各位党员大家好:这天我能站在鲜红的党旗下,
【发言稿】 日期:2022-12-24
-
破冰提能大讨论个人发言4篇
破冰提能大讨论个人发言4篇破冰提能大讨论个人发言篇1党史学习教育开展以来,我坚持读原著、学原文、悟原理。今天,根据会议安排,现在我就“学史明理”主题谈几点个
【发言稿】 日期:2022-10-09
-
巡察整改专题民主生活会总结发言8篇
巡察整改专题民主生活会总结发言8篇巡察整改专题民主生活会总结发言篇1按照区委统一部署和纪监委、巡察办关于召开党史学习教育专题组织生活会的工作安排,近期我紧贴
【发言稿】 日期:2022-10-12
-
我最敬佩的人开头_我敬佩的一个人作文20篇2020年
我敬佩的一个人作文20篇 我敬佩的一个人作文一): 我身边有很多值得我们敬佩的人,但我最敬佩的一
【发言稿】 日期:2020-11-10
-
学习周永开先进事迹心得体会3篇
学习周永开先进事迹心得体会【一】通过学习周永开老先生先进事迹后,结合自己工作思考,感慨万千。同样作为
【格言】 日期:2021-04-10
-
最满意的三项工作200字【最新党办公务员副主任提拔考察个人三年思想工作总结报告】
党办公务员个人三年工作总结近三年来,本人在组织、领导的关心指导和同事们的团结协作下,尽快完成主角的转
【格言】 日期:2021-02-26
-
XX老干局推进党建与业务深度融合发展工作情况调研报告:党建调研报告
XX老干局推进党建与业务深度融合 发展工作情况的调研报告 党建工作与业务工作融合发展始终是一个充满生
【成语大全】 日期:2020-08-28
-
中国共产党第三代中央领导集体的卓越贡献
中国共产党第三代中央领导集体的卓越贡献 --------------继往开来铸就辉煌 【摘要】改
【成语大全】 日期:2020-03-20
-
信息技术2.0能力点 [全国中小学教师信息技术应用能力提升工程试题题库及参考答案「精编」]
全国中小学教师信息技术应用能力提升工程试题题库及答案(复习资料)一、判断题题库(A为正确,B为错误)
【格言】 日期:2020-11-17
-
党建工作运行机制内容有哪些_构建基层党建工作运行机制探讨
党的基层组织是党在社会基层组织中的战斗堡垒,是党的全部工作和战斗力的基础。加强和改进县级以下各类党的
【经典阅读】 日期:2020-01-22
-
电大现代教育原理_最新国家开放大学电大《现代教育原理》形考任务2试题及答案
最新国家开放大学电大《现代教育原理》形考任务2试题及答案形考任务二一、多项选择题(共17道试题,共3
【成语大全】 日期:2020-07-20
-
集合推理_七,推理与集合
七推理与集合1 期中考试数学成绩出来了,三个好朋友分别考了88分,92分,95分。他们分别考了多少分
【名人名言】 日期:2020-12-18
-
基层党务工作基本内容_党建基本工作有哪些
党建基本工作有哪些(一) 基层党建工作包括哪些内容 选择了大学生村官这条路,你就与农村基层党
【名人名言】 日期:2020-08-06
-
[人生赠言]城隍庙人生赠言注释
人之相惜惜于品,人之相敬敬于德,人之相交交于情,人之相随随于义,人之相拥拥于礼,人之相信信于诚,人之相伴伴于爱! 人生的确很累,看你如何品味,每天多寻欢乐,烦...
【格言】 日期:2019-07-14
-
关于三农工作重要论述心得体会3篇
关于三农工作重要论述心得体会3篇关于三农工作重要论述心得体会篇1习近平总书记指出:“建设现代化国家离不开农业农村现代化,要继续巩固脱贫攻坚成果,扎实推进乡村
【学习心得体会】 日期:2022-10-29
-
【福生庄隧道坍塌处理方案】 福生庄隧道在哪里
(呼和浩特铁路局大包电气化改造工程指挥部,内蒙古呼和浩特010050)摘要:文章介绍了福生庄隧道
【学习心得体会】 日期:2020-03-05
-
五个一百工程阅读心得体会13篇
五个一百工程阅读心得体会13篇五个一百工程阅读心得体会篇1凡益之道,与时偕行。在全国网络安全和信
【学习心得体会】 日期:2022-12-07
-
城管系统警示教育心得体会9篇
城管系统警示教育心得体会9篇城管系统警示教育心得体会篇1各党支部要召开多种形式的庆七一座谈会,组织广大党员进行座谈,回顾党的光辉历程,畅谈党的丰功伟绩,
【学习心得体会】 日期:2022-10-09
-
发展对象培训主要内容10篇
发展对象培训主要内容10篇发展对象培训主要内容篇1怀着无比激动的心情,我有幸参加了__新区区委党校20__年第四期(区级机关)党员发展对象培训班。这次的学习
【培训心得体会】 日期:2022-09-24
-
扶眉战役纪念馆心得体会11篇
扶眉战役纪念馆心得体会11篇扶眉战役纪念馆心得体会篇1有那么一段历史,低诉着血和泪的故事,慢慢地,随岁月老去;有那么一群人,放弃了闲逸的人生,辗转奔波中
【学习心得体会】 日期:2022-08-03
-
凝聚三种力量发展全过程人民民主心得体会12篇
凝聚三种力量发展全过程人民民主心得体会12篇凝聚三种力量发展全过程人民民主心得体会篇1新民主主义革命是指在帝国主义和无产阶级革命时代,殖民地半殖民地国家中的
【学习心得体会】 日期:2022-08-31
-
2022年全国检察长会议心得7篇
2022年全国检察长会议心得7篇2022年全国检察长会议心得篇1眼睛是心灵上的窗户,我们通过眼睛才能看到世间万物,才能看到眼前这美好的一切。拥有一双明亮的眼
【学习心得体会】 日期:2022-10-31
-
全面从严治党的心得体会800字7篇
全面从严治党的心得体会800字7篇全面从严治党的心得体会800字篇1中国特色社会主义是我们党领导
【学习心得体会】 日期:2022-12-14
-
教师全国两会精神学习专题研讨交流材料6篇
教师全国两会精神学习专题研讨交流材料6篇教师全国两会精神学习专题研讨交流材料篇1通过对两会精神深入系统的学习,作为新一代的青年人,更应该严格要求自己,贯彻落
【教师心得体会】 日期:2022-08-11
-
 2024年主题教育民主生活会批评与自我批评意见(38条)(范文推荐)
2024年主题教育民主生活会批评与自我批评意见(38条)(范文推荐)
2023年主题教育民主生活会六个方面个人检视、相互批评意见:1 理论学习系统性不强。学习习近平新时代中国特色社会主义思想不深不透,泛泛而学的时候多,深学细照的时候少,特...
【邓小平理论】 日期:2024-03-19
-
 2024年交流发言:强化思想理论武装,增强奋进力量(完整)
2024年交流发言:强化思想理论武装,增强奋进力量(完整)
习近平总书记指出:“一个民族要走在时代前列,就一刻不能没有理论思维,一刻不能没有思想指引。”党的十八大以来,伴随着新时代中国特色社会主义思想在实践中形成发展的历程...
【三个代表】 日期:2024-03-19
-
2024年度镇年度县乡人大代表述职评议活动总结
xx镇20xx年县乡人大代表述职评议活动总结为响应县级人大常委会关于开展县乡两级人大代表述职评议活动,进一步激发代表履职活力,加强代表与人民群众的联系,提高依法履职水平...
【马克思主义】 日期:2024-03-19
-
 “千万工程”经验学习体会(研讨材料)
“千万工程”经验学习体会(研讨材料)
“千万工程”是总书记在浙江工作时亲自谋划、亲自部署、亲自推动的一项重大决策,也是习近平新时代中国特色社会主义思想在之江大地的生动实践。20年来,“千万工程”先后经历...
【三个代表】 日期:2024-03-19
-
 2024年在市政协机关工作总结会议上讲话
2024年在市政协机关工作总结会议上讲话
同志们:刚才,XX同志对市政协机关20XX年工作进行了很好的总结,很精炼,很到位,可以感受到去年机关工作确实可圈可点。XX同志宣读了表彰决定,机关优秀人员代表、先进集体代...
【邓小平理论】 日期:2024-03-18
-
 在全区防汛防涝动员暨河长制工作推进会上讲话提纲【完整版】
在全区防汛防涝动员暨河长制工作推进会上讲话提纲【完整版】
区长,各位领导,同志们:汛期已经来临,我区城区防涝工作面临强大考验,形势不容乐观。年初,区城区防涝排渍指挥部已经召开专题调度会,修订完善应急预案,建立网格化管理机...
【马克思主义】 日期:2024-03-18
-
 2024年镇作风整治工作实施方案(完整文档)
2024年镇作风整治工作实施方案(完整文档)
XX镇作风整治工作实施方案为深入贯彻落实党的二十大精神及省市区委深化作风建设的最新要求,突出重点推进干部效能提升,坚持不懈推动作风整治工作纵深发展,根据《关于印发《2...
【毛泽东思想】 日期:2024-03-18
-
2024市优化法治化营商环境规范涉企行政执法实施方案【优秀范文】
xx市优化法治化营商环境规范涉企行政执法实施方案为持续优化法治化营商环境,激发市场主体活力和社会创造力,规范行政执法行为,创新行政执法方式,提升行政执法质效,着力解...
【毛泽东思想】 日期:2024-03-18
-
 2024年度关于开展新一轮思想状况摸底排查工作通知(完整)
2024年度关于开展新一轮思想状况摸底排查工作通知(完整)
关于开展新一轮思想状况摸底排查工作的通知为深入贯彻落实关于各地开展干部职工思想状况大摸底大排查情况上的批示要求和改革教育第二次调度会议精神,有针对性做好队伍教育管...
【三个代表】 日期:2024-03-18
-
2024年公路养护中心主任典型事迹材料(完整文档)
“中心的工作就是心中的事业”——公路养护中心主任典型事迹材料**,男,1976年6月出生,1993年参加工作,2000年4月调入**区交通运输局工作,大学本科学历,中共党员,现任**...
【马克思主义】 日期:2024-03-17